Оглавление
Синдром элерса-данло (ehlers-danlos) (сэд; q79. 6) — генетически гетерогенное заболевание, обусловленное разнообразными мутациями в генах коллагена, либо в генах, отвечающих за синтез ферментов, принимающих участие в созревании волокон коллагена.
Синдром Элерса-Данло. Причины. Симптомы. Диагностика. Лечение

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Элерса-Данло (Ehlers-Danlos) (СЭД; Q79.6) — генетически гетерогенное заболевание, обусловленное разнообразными мутациями в генах коллагена, либо в генах, отвечающих за синтез ферментов, принимающих участие в созревании волокон коллагена.
Источник
«Мои особенности делают меня лишь сильнее» — Wonderzine
Диагноз и сериал «Кости»
Синдром Элерса — Данлоcа (СЭД) — это генетическое заболевание соединительной ткани. Его вызывают дефекты в синтезе коллагена — важнейшего белка мышц, кровеносных сосудов, костей, хрящей и сухожилий, наружных покровов всех органов. Коллаген «склеивает» ткани, придавая им силу и эластичность — «ломаный» коллаген не выполняет свои обязанности. Этот же белок в виде ампул или инъекций не помогает: организм не знает, что такое «нормальный» коллаген, не способен его вырабатывать и усваивать. Связки не поддерживают суставы — из-за этого происходят вывихи. Позвоночник не фиксируется на месте — в результате появляется искривление. Чаще всего людей с этим синдромом объединяет хрупкость сосудов, повышенная гибкость суставов и невероятная эластичность кожи, почти как у Мистера Фантастика. А ещё — постоянная боль.
С детства у меня болит спина. Родители водили меня к лучшим хирургам, ортопедам, остеопатам, но все говорили: «У вас искривление позвоночника, ходите на плавание». Без достаточных обследований мне назначали лечение, которое только вредило. Например, физические упражнения не учитывающие, что у меня есть протрузии (это первая стадия формирования грыж межпозвонковых дисков), — а они могут привести к грыжам. Или массажи, стимулирующие приток крови к пояснице, что запрещено при эндометриозе (о том, что у меня есть и он, я узнала случайно всего полгода назад — хотя регулярно на протяжении многих лет посещала гинекологов и упоминала боли и другие симптомы). Практически каждый врач упрекал, меня или родителей, что мы не уследили за моим здоровьем и довели до такого состояния. Однажды хирург в районной больнице на вопрос, что мне делать со спиной, ответил: «Ничего. Ждите годы боли».
Осенью 2016 года я оказалась на приёме у одного из лучших вертебрологов (специалист по заболеваниям позвоночника. — Прим. ред.) Латинской Америки. Я встречалась с мексиканцем и часто к нему летала; он знал, что у меня всё время болит спина, а в России я не могу добиться обследования, и записал к специалисту. За два дня там я узнала о своём состоянии больше, чем за двадцать лет здесь. После осмотра и нескольких рентгеновских снимков вертебролог составил список обязательных обследований: магнитно-резонансная томография, компьютерная томография и другие.
Всё выяснилось год назад совершенно случайно, благодаря сериалу «Кости».
Там у одной из жертв был синдром
Элерса — Данлоса — и такие же симптомы, как у меня
Вернувшись в Москву, я три месяца пыталась получить направление на МРТ. Когда невролог назвал опытнейшего мексиканского вертебролога дураком, моё терпение лопнуло. Я пошла к заведующей больницы, но она сказала, что я никогда не получу направление на МРТ позвоночника, потому что им запрещено назначать такой объёмный анализ. Только после того, как я без сил разрыдалась, она сжалилась надо мной и дала направление на томографию одного отдела позвоночника, хотя мне нужно было обследовать четыре. После этого я никогда больше не пользовалась бесплатной медициной.
Всё выяснилось год назад совершенно случайно, благодаря сериалу «Кости». Там у одной из жертв был синдром Элерса — Данлоса — и такие же симптомы, как у меня. Я удивилась сходству, почитала о болезни в интернете и записалась к генетику в Москве — единственному российскому специалисту, который посещает международные конференции, посвящённые СЭД. Я пришла на приём с папкой анализов и заключений всех возможных специалистов. Врач их изучила, спросила о заболеваниях родственников, осмотрела меня и подтвердила моё предположение, поставив диагноз «синдром Элерса — Данлоса». С одной стороны я испытала облегчение: наконец я знаю, что со мной, мне будет легче контролировать болезнь. С другой — диагноз звучал как приговор, поскольку СЭД неизлечим. Генетик утешила, что с возрастом гипермобильность уменьшится. Но, к сожалению, другие хронические болезни, появившиеся из-за синдрома, никуда не денутся.

Источник
Статья посвящена проблеме тробогеморрагических осложнений во время беременности и родов у больных с врожденными заболеваниями соединительной ткани
Тробогеморрагические осложнения во время беременности и родов у больных с врожденными заболеваниями соединительной ткани
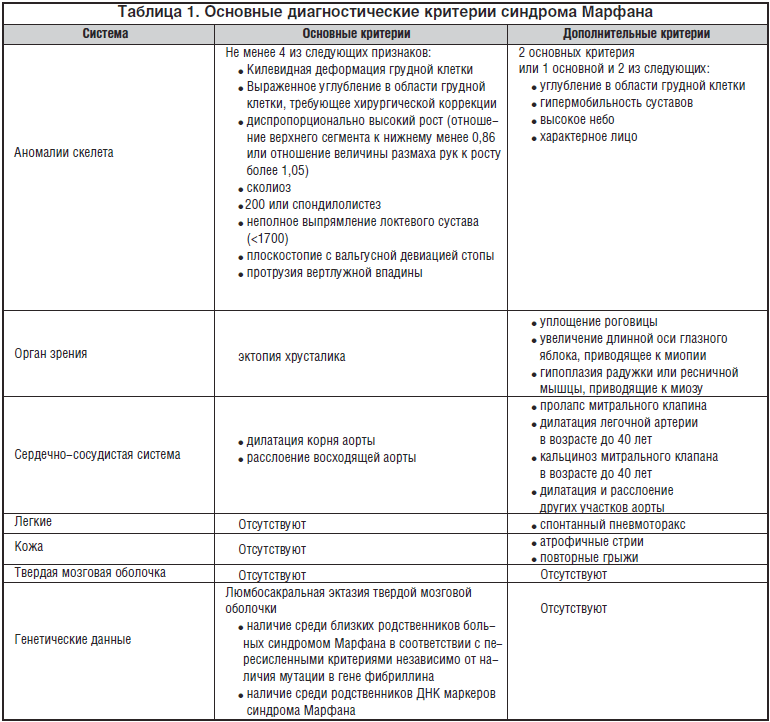
Синдром Марфана представляет собой аутосомно-доминантное заболевание соединительной ткани, связанное с мутацией в гене фибриллина — одного из основных эластических компонентов соединительной ткани [5,7,14]. При этом нарушается синтез микрофибриллярных волокон в стенках сосудов, клапанах сердца, связках, суставах, твердой мозговой оболочке, костях и других органах. Распространенность синдрома Марфана, по данным различных источников, составляет 1 на 5-10 тыс. населения [5,14,18,20]. Наиболее частыми клиническими признаками заболевания являются прогрессирующая дилатация аорты, пролапс и недостаточность митрального и аортального клапанов, гипермобильность суставов, высокий рост, длинные конечности, арахнодактилия, деформация грудной клетки, сколиоз, эктопия хрусталика, миопия [5,8,18]. Особое значение имеют проявления со стороны сердечно-сосудистой системы, так как именно с ними связана высокая летальность при этом заболевании [18]. Молекулярная диагностика синдрома Марфана проводится редко, в связи с чем в настоящее время согласно международным рекомендациям используется клиническая диагностика в соответствии с диагностическими критериями Ghent, предложенными в 1996 году взамен ранее существовавших Берлинских критериев 1988 года (табл. 1).
Для постановки диагноза необходимо по 1 основному критерию со стороны 2 различных систем органов (4 признака со стороны скелета составляют 1 основной критерий) и 1 дополнительный критерий.
При ведении беременных с синдромом Марфана следует учитывать два основных обстоятельства:
1. Беременные с синдромом Марфана имеют высокий риск развития летальных осложнений со стороны сердечно-сосудистой системы (разрыва аорты и расслоения аневризмы аорты).
2. Заболевание наследуется у детей в 50% случаев.
Больным с синдромом Марфана показано тщательное обследование в период планирования беременности, обязательно включающее трансторакальное или трансэзофагеальное ультразвуковое исследование аорты. При диаметре аорты более 4 см беременность противопоказана. Все необходимые хирургические вмешательства на клапанах и аорте больным с синдромом Марфана должны быть произведены до беременности. Женщины должны быть предупреждены о возможных летальных осложнениях во время беременности и о риске наследования заболевания ребенком. Кроме того, необходимо информировать пациентку и ее родственников о снижении продолжительности жизни в связи с беременностью и ухудшении течения заболевания. Риск наследования синдрома Марфана плодом можно определить уже в конце 1 триместра при генетическом исследовании.
Большинство осложнений со стороны сердечно-сосудистой системы развивается во втором или третьем триместрах беременности, хотя описаны случаи расслоения аорты на ранних сроках беременности, в родах и на протяжении 8 дней послеродового периода. Причинами высокого риска расслоения аневризмы аорты во время беременности у пациенток синдромом Марфана являются физиологическое увеличение объема циркулирующей крови и сердечного выброса на фоне врожденной аномалии коллагена. Определенную роль также имеют гормональные изменения. Гестационная гипертензия и гестозы резко увеличивают риск дилатации, расслоения и разрыва аорты. Своевременная диагностика и терапия внезапного расслоения аневризмы аорты жизненно необходимы, так как около 50% пациентов погибает в течение 48 часов после возникновения осложнения. При беременности этот процент значительно выше. Основными симптомами являются боль в груди, иррадиирующая в спину, плечи и живот. Другие симптомы связаны с осложнениями данного состояния. Наиболее опасными осложнениями являются кровотечения в перикард, плевральную полость, средостение, забрюшинное пространство, стенку легочной артерии, полости сердца. Кроме того, нередко наблюдаются симптомы, связанные с частичной или полной окклюзией различных артерий гематомой средней оболочки аорты. Окклюзия коронарных артерий может привести к внезапной смерти или инфаркту миокарда, общей сонной — к синкопальным состояниям, инсульту или коме, подключичной артерии—ишемии верхних конечностей и парезам, межреберных или поясничных артерий — ишемии спинного мозга. Вследствие дилатации или расслоении аорты на уровне аортального клапана может развиться выраженная аортальная недостаточность и отек легких. Обструкция аорты или легочной артерии нередко приводит к циркуляторному коллапсу.
При физическом обследовании часто выявляются дефицит пульса, диастолический шум на аорте, неврологические проявления (цереброваскулярные нарушения, потеря сознания, парапарезы или параплегии). При рентгенографии грудной клетки обнаруживается расширение средостения. Иногда имеются признаки гемоторакса (в основном левостороннего при расслоении дистального участка аорты). Однако данные рентгенографии неспецифичны и отсутствие патологических изменений на рентгенограммах не позволяет исключить диагноз. Золотым стандартом диагностики расслоения аорты является аортография. Однако во время беременности методами выбора являются контрастная высокоразрешающая компьютерная томография, магнитно-резонансное исследование, трансэзофагеальная эхокардиография и ультразвуковое исследование, что связано с их неинвазивностью и отсутствием отрицательного влияния на плод.
Дифференциальный диагноз расслоения аневризмы аорты у беременных с синдромом Марфана проводится с такими острыми состояниями, как эмболия околоплодными водами, инфаркт миокарда и аортальная регургитация, вызванные другими причинами, а также пневмотораксом, инсультом, разрывом матки, отслойкой плаценты, тромбозом мезентериальных сосудов. В большинстве случаев диагноз расслоения аневризмы аорты устанавливается post mortem.
Для предотвращения и своевременной коррекции угрожающих жизни осложнений на протяжении всей беременности больные с синдром Марфана должны находиться под тщательным наблюдением акушеров и сосудистых хирургов. Всем беременным с синдромом Марфана (даже не ранее не имевшим признаков поражения сердечно-сосудистой системы) показано трансторакальное ультразвуковое исследование или МРТ в динамике. Во многих работах доказана эффективность b-адреноблокаторов для предотвращения прогрессирующей дилатации аорты, аортальной регургитации, расслоения аневризмы аорты, развития застойной сердечной недостаточности [8,14]. При терапии b-адреноблокаторами следует учитывать их возможные побочные эффекты, включающие задержку развития плода, брадикардию, гипогликемию, гипербилирубинемию, повышение тонуса матки, апноэ у новорожденных. В связи с секрецией b-адреноблокаторов в грудное молоко кормление следует проводить только через 3-4 часа после приема препарата, когда его концентрация в плазме начинает снижаться. Четких рекомендаций о показаниях к профилактическому назначению b-адреноблокаторов во время беременности больным синдромом Марфана на данный момент не существует. При расслоении аневризмы дистального отдела аорты используется внутривенное введение b-адреноблокаторов до достижения снижения ЧСС на 20%, снижения АД и сократимости левого желудочка. В последующем переходят на поддерживающую дозу оральных b-адреноблокаторов. Необходимо снижение систолического АД до 100—120 мм рт.ст. или до минимального уровня, необходимого для кровоснабжения жизненноважных органов. Основным средством для снижения АД вне беременности является нитропруссид натрия, обладающий рядом преимуществ по сравнению с другими препаратами (быстрое начало воздействия, короткий период полувыведения и легкость в подборе необходимой дозы). Однако в связи с возможным токсическим действием на плод его применение у беременных ограничено. Поэтому средством выбора для снижения АД у беременных с синдромом Марфана является гидралазин в комбинации с b-адреноблокаторами. Хирургическое вмешательство при расслоении дистального отдела аорты показано при неэффективности медикаментозной терапии, разрыве или угрозе разрыва аорты, прогрессирующем расслоении аневризмы аорты. При расслоении аневризмы проксимального отдела аорты необходимо срочное оперативное вмешательство, так как только эта мера способна предотвратить летальный исход. Срочное хирургическое вмешательство показано также беременным с синдромом Марфана при увеличении диаметра аорты свыше 45 мм: на ранних сроках рекомендуется прерывание беременности, на поздних—кесарево сечение с последующей реконструктивной операцией на аорте. Другим частым показанием к неотложному оперативному вмешательству во время беременности у больных с синдромом Марфана является прогрессирующая аортальная недостаточность. Успех оперативных вмешательств у беременных с синдромом Марфана зависит от тяжести осложнения, срока беременности, своевременности вмешательства. В большинстве случаев хирургическое лечение приводит к прерыванию беременности, поэтому при жизнеспособном плоде до или одновременно с операцией на сердце и сосудах проводится кесарево сечение.
Оптимальным методом родоразрешения беременных с синдромом Марфана является кесарево сечение, что позволяет минимизировать гемодинамические изменения, связанные с вагинальным родоразрешением. Лишь в немногих случаях при диаметре аорты менее 40 мм, отсутствии сопутствующих проявлений заболевания, адекватном обезболивании и хорошем контроле артериального давления возможны роды через естественные родовые пути. В определенных случаях для укорочения второго периода родов показаны акушерские щипцы. Некоторые авторы [8,18] рекомендуют одновременно с кесаревым сечением производить гистерэктомию, так как в послеродовом периоде у родильниц с синдромом Марфана часто отмечаются массивные маточные кровотечения. Причиной таких кровотечений является нарушение сократительной способности спиральных артерий, что имеет место и при других заболеваниях соединительной ткани (например, при синдроме Элерса-Данло).
Синдром Элерса-Данло
Синдром Рендю-Ослера
Синдром Рендю-Ослера (врожденная геморрагическая телеангиоэктазия) представляет собой группу аутосомно-доминантных заболеваний. Распространенность заболевания, по различным данным, составляет от 5 до 8 на 1000 населения [6,17]. Клинические проявления наследственной геморрагической телеангиоэктазии связаны с нарушением структуры стенки сосудов (рис. 1).
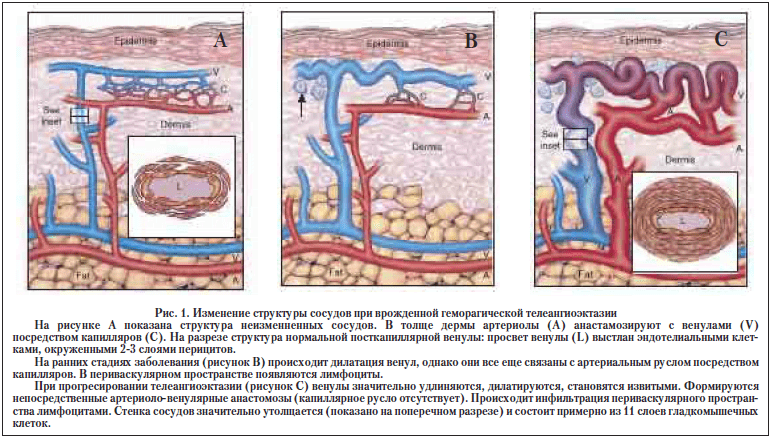
Наиболее частым клиническим проявлением синдрома Рендю-Ослера являются спонтанные рецидивирующие носовые кровотечения (имеются в 78-96% случаев). Другим частым симптомом являются телеангиоэктазии на коже и слизистых, которые появляются в более позднем возрасте, чем носовые кровотечения и имеют тенденцию увеличиваться в размерах и количестве с возрастом. Телеангиоэктазии располагаются на губах, языке, мягком небе, слизистой полости рта, лице, конъюнктивах, ногтевых ложах, пальцах. Более редким проявлением заболевания являются артерио-венозные мальформации, расположенные в различных органах: легких, ЖКТ, печени, головном и спинном мозге. Большую опасность представляют легочные артерио-венозные мальформации. Это прямые анастомозы между легочным артериальным и венозным руслом, минуя капилляры. Симптомы легочных артерио-венозных мальформаций включают в себя одышку, цианоз, кашель, боль в области грудной клетки и др. Наличие легочных артерио-венозных мальформаций может привести к ряду серьезных осложнений, которые можно предотвратить своевременной диагностикой и лечением. Наиболее частыми являются неврологические осложнения, которые включают в себя инсульты, транзиторные ишемические атаки, абсцессы мозга, мигрени. Более редкими, но крайне опасными осложнениями являются гемоторакс и легочное кровотечение.
Основными методами диагностики легочных артерио-венозных мальформаций являются рентгенологическое исследование грудной клетки, методика со 100% кислородом, контрастная эхокардиография, сцинтиграфия, компьютерная томография. Основным методом лечения является чрескожная эмболотерапия.
Иногда у пациентов с врожденной геморрагической телангиэктазией имеются нарушения в системе гемостаза. У многих из них выявляются дефекты функции тромбоцитов и системы фибринолиза. Примерно у 50% больных имеется нерезковыраженный ДВС-синдром, редко переходящий в фульминантную форму.
Таким образом, больные с синдромом Рендю-Ослера имеют высокий риск развития угрожающих жизни осложнений, для предотвращения которой чрезвычайно важна своевременная диагностика заболевания. Для стандартизации подходов к диагностике и для предотвращения случаев гипердиагностики на последнем международном конгрессе в 2000 году были выработаны следующие диагностические критерии заболевания (табл. 2). 
Все потомство больных наследственной геморрагической телеангиоэктазией имеет потенциальный риск манифестации заболевания в более позднем возрасте. При постановке диагноза следует исключить дефекты гемостаза. При наличии висцеральных проявлений заболевания у детей следует тщательно проверить остальных членов семьи. В будущем, вероятнее всего, клинический диагноз в соответствии с вышеперечисленными критериями будет заменен молекулярными тестами, которые станут общедоступными.
Беременность у женщин с болезнью Рендю-Ослера связана с чрезвычайно высоким риском осложнений. Вопрос о беременности должен решаться индивидуально в каждом конкретном случае, однако благоприятный исход беременности следует ожидать лишь при правильном ее ведении, включающем своевременную диагностику и терапию осложнений заболевания. Большую опасность представляют осложнения, связанные с наличием легочных артерио-венозных мальформаций, в частности, массивные легочные кровотечения. Изменение гемодинамики и гормонального статуса беременных приводит к ухудшению состояния ранее существовавших мальформаций и росту новых. Во время беременности объем циркулирующей крови увеличивается на 40%, сердечный выброс — на 30-50%, что способствует значительному повышению кровотока в легких и, как следствие, дилатации или разрыву тонкостенных сосудов артерио-венозных мальформаций. Чаще подобные осложнения имеют место во 2-3 триместрах беременности, когда увеличение объема циркулирующей крови и сердечного выброса достигают максимума. Кроме того, высокий уровень прогестерона во время беременности способствует ослаблению стенок венозных сосудов, что также приводит к росту артерио-венозных мальформаций.
На данный момент не существует четких рекомендаций о наиболее целесообразном методе родоразрешения женщин с наследственной геморрагической телеангиоэктазией. Отсутствуют исследования о сравнительной безопасности консервативного и оперативного родоразрешения. В большинстве наблюдений различных авторов [6,17] беременность у женщин с наследственной геморрагической телеангиоэктазией завершилась плановым или экстренным кесаревым сечением. Случаи экстренного оперативного родоразрешения были связаны в основном с несвоевременной диагностикой и коррекцией угрожающих жизни осложнений, (в частности, легочных кровотечений и гемоторакса) [17], причем исход для плода в подобных случаях был не всегда благоприятным и зависел в основном от срока родоразрешения. Плановое оперативное родоразрешение, по-видимому, показано беременным с имеющимися легочными или церебральными мальформациями (или подозрением на их наличие) для уменьшения риска разрыва мальформаций и кровотечений во время родов через естественные родовые пути.
Консервативное родоразрешение беременных с наследственной геморрагической телеангиоэктазией следует рекомендовать при отсутствии церебральных и легочных мальформаций или при их полноценной коррекции до беременности.
В связи с возможными артерио-венозными мальформациями спинного мозга таким беременным не рекомендуется проведение эпидуральной анестезии. Для исключения мальформаций спинного мозга показано магнитно-резонансное исследование.
Таким образом, наиболее безопасной стратегией для женщин с наследственной геморрагической телеангиоэктазией является планирование беременности с тщательным обследованием до беременности, в частности, на предмет наличия артерио-венозных мальформаций в легких и головном мозге (ангиография, МРТ, КТ) и при необходимости их хирургической коррекции. Кроме того, следует учитывать, что риск подобной патологии у детей составляет 50%.
Мы имеем опыт ведения беременности и родов у больных с врожденными заболеваниями соединительной ткани. За последние 10 лет в родильном доме при 67 ГКБ г. Москвы мы наблюдали 27 беременных с врожденными заболеваниями соединительной ткани (12 с синдромом Марфана, 9 с синдромом Элерса-Данло и 6 с синдромом Рендю-Ослера).
Большинство беременных с врожденными заболеваниями соединительной ткани имели различные осложнения во время беременности, родов и послеродового периода, включая дилатацию восходящей аорты, развитие аортальной недостаточности, тромбоз левого предсердия, увеличение выраженности телеангиоэктазий, прогрессирование носовых, десновых или желудочно-кишечных кровотечений, появление артерио-венозных мальформаций в легких, тяжелые послеродовые кровотечения, развитие гематом послеоперационного шва. У 22 беременных с врожденными заболеваниями соединительной ткани выявлены различные дефекты в системе гемостаза, включая нарушение функции тромбоцитов, дефекты в системе фибринолиза и признаки ДВС-синдрома. 24 из 27 беременных с врожденными заболеваниями соединительной ткани были родоразрешены путем операции кесарево сечение, причем основным показанием к операции во всех случаях явилась соматическая патология. Во время операции у большинства таких больных отмечалась повышенная кровоточивость из сосудов мелкого и среднего калибра. С целью профилактики и коррекции тромбогеморрагических осложнений использовались свежезамороженная плазма, транексамовая кислота.
Заключение
Таким образом, беременность у больных с врожденными заболеваниями соединительной ткани сопровождается высоким риском различных осложнений, наиболее опасными из которых являются тромбогеморрагические осложнения. В последние годы появились методы диагностики, позволяющие детально обследовать таких больных и своевременно выявлять осложнения, благодаря современным фармакологическим препаратам возможно значительно снизить летальность от тромбогеморрагических осложнений и обеспечить относительно безопасное ведение беременности и родов у данной категории пациенток. Для благоприятного исхода беременности у больных с врожденными заболеваниями соединительной ткани необходимо знание особенностей заболеваний врачами различных специальностей, а также тесная кооперация кардиологов, акушеров, неонатологов и сосудистых хирургов, начиная с момента планирования беременности.
Источник
Cиндром Элерса-Данлоса в Германии: предотвращение прогрессирования заболевания, лечение симптомов. Комплексное лечение и диагностика синдорома Элерса-Далоса
Cиндром Элерса-Данлоса: лечение
Лечение синдрома Элерса-Данло в Германии направлено на предотвращение прогрессирования заболевания, лечение симптоматики и проходит под постоянным наблюдением врачей. Качество жизни пациентов часто ограничивается, однако, немецкая медицина делает все, чтобы улучшить состояние пациента и поддерживать функционирование всего организма в норме. Эти пациенты получают терапию, направленную на предотвращение получения травм суставов и против плохого заживления ран.
Также при тяжелых формах в клиниках Германии по лечению редких заболеваний проводят операции. Комплексное лечение синдорома Элерса-Далоса в Германии приводит к хорошим прогнозам для пациента.
Редкие заболевания
Клиники
Новости



- Лучше клиники Германии
- Только профильные врачи
- Индивидуальный подход
- Быстрая обратная связь
- Врачебная тайна
- Взаимодействие с клиентом
- Клиентская поддержка 24/7
- Возврат платежа в случае отказа клиники
- Юридическая ответственность врача
- Медицинский перевод выписок
- Независимое мнение нескольких врачей
- 100% конфиденциальность
Какие виды лечения в Германии?
Немецкая медицинская служба опеки EMEX Medical GmbH предоставляет максимально широкий спектр услуг, касающихся лечения в Германии. Мы сотрудничаем с клиниками, которые специализируются на лечении самых разных направлений: кардиология, гастроэнтерология, офтальмология, хирургия, онкология, ортопедия. Помимо этого, лечение в Германии доступно не только взрослым, но и детям – с учетом всех возрастных особенностей. Конечно же, перед началом лечения необходимо пройти диагностику – с этим профессионально справятся немецкие врачи, при помощи новейших диагностических процедур и технологий.
Если Вы хотите не только получить высококачественное лечение и диагностику в Германии, но и провести для себя профилактические меры, связанные с предотвращением возможных осложнений к болезням, к которым у Вас есть предрасположенность – обращайтесь к команде профессионалов EMEX Medical.
Остались вопросы? Консультанты нашей службы с радостью предоставят всю необходимую информацию, касающуюся лечения в Германии, а также организационных процедур. Для этого воспользуйтесь кнопкой обратной связи, размещенной на нашем сайте.
Источник


