Оглавление
- 1 Сколько хромосом у свиньи?
- 2 Методы исследования хромосом
- 3 Немного о РНК
- 4 Проблема терминологии
- 5 Предрасположенность = болезнь?
- 6 Как паре, планирующей ребенка, понять, что нужно идти к врачу-генетику?
- 7 Как расшифровать результат анализа
- 8 Что делать, если хромосомный анализ показал вероятность рождения больного ребенка
Не все семьи могут проследить все возможные рецессивные заболевания. Тогда на помощь приходит экзомное секвенирование — тест для определения генетических повреждений (мутаций) в ДНК путем изучения в одном тесте практически всех участков генома, кодирующих белки, изменения которых являются причиной наследственных заболеваний.
Сколько хромосом у свиньи?
Домашняя свинья — крупный парнокопытный подвид дикого кабана. Он был одомашнен человеком более 7 тысяч лет назад и широко распространен в странах Восточной Азии, на Западе и в Океании. Сегодня дикие свиньи — рейнджеры — часто встречаются в Австралии, Северной Америке и Новой Зеландии. Взрослая свинья весит от 50 до 150 кг в зависимости от вида, а длина ее тела составляет от 0,9 до 1,8 м. Животные, живущие в дикой природе, обычно травоядные (кроме кабана). Но, те виды, которые одомашнены человеком, всеядны.
Со школьных лет мы знаем, что человек произошел от обезьяны. Никто не помнит, как это было на самом деле, но все повторяют слова Дарвина. Существуют теории о внеземном происхождении человека, в том числе и о Божественном, но о метафизике мы сейчас говорить не будем.

В июне не было новых сообщений в блоге, я занимался научным исследованием теории генезиса свиней. Пока окончательно не решено, где публиковать первые результаты, в «Ланцете» или в «Медицинском журнале Новой Англии», так что вот что я вам скажу.
После долгих размышлений я пришел к выводу, что человек произошел от свиньи. Теория обезьян — ложь! Свинья ближе к человеку не только биологически, но и социокультурно.
Мы с тобой одной крови
На самом деле обезьян всегда называли нашими ближайшими родственниками. Сейчас наука пытается доказать, что семейство свиней нам ближе всего. Однако это не означает, что человек произошел от свиньи.
Любое животное может претендовать на эту должность. Клетки ДНК человека по дезоксирибонуклеиновой кислоте совпадают почти со всеми видами млекопитающих. Различия с шимпанзе в генах составляют 1-2%. Сколько матчей со свиньями пока неизвестно. Но уже было показано, что его кровь практически полностью соответствует человеческой. Молекулы гормона роста похожи на 70%.
Поражает элементарное сравнение внутреннего веса человека и свиньи: сердце человека весит 300 г, сердце свиньи весит на 20 г больше, легкие весят соответственно 790 г и 800 г, такие же близкие показатели для почек и печень.
По словам Екатерины, это свидетельствует о том, что, несмотря на тяжесть состояния и высокую смертность, считать синдром Эдвардса смертельным заболеванием, несовместимым с жизнью, некорректно: «Моя дочь растет, развивается, она личность и только личность девушка», хотя и не нормотипичная, но со своим характером и потребностями».
Методы исследования хромосом
Для изучения кариотипа используют специальный метод — световую микроскопию дифференциально окрашенных метафазных хромосом культивируемых лимфоцитов периферической крови.
Этот анализ используется для диагностики различных хромосомных заболеваний. Он позволяет выявлять такие нарушения, как:
- Большие изменения кариотипа: изменение числа хромосом. Например, при синдроме Дауна в клетках ребенка присутствует дополнительная хромосома 21.
- Наличие в организме клеток с разным кариотипом. Это явление называется мозаицизмом.
- Хромосомные аберрации – нарушение строения хромосом, внутрихромосомные и межхромосомные перестройки. К ним относятся делеции (потеря участка хромосомы), дупликации (удвоение участка хромосомы), инверсии (поворот участка хромосомы на 180 градусов), транслокации (перенос участка одной хромосомы на другую).
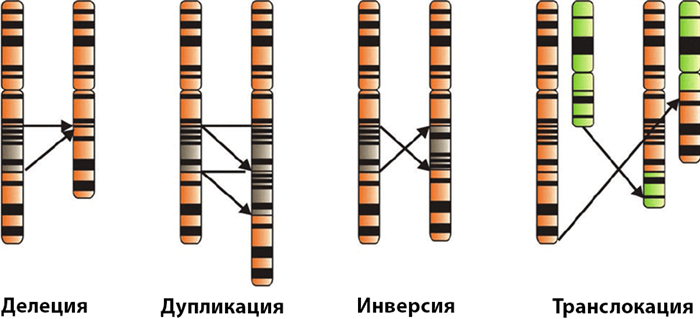
Однако не все генетические нарушения можно выявить с помощью исследования кариотипа. Он не может обнаружить такие изменения, как:
- микроделеции и микродупликации, когда теряется или дублируется очень небольшой участок хромосомы;
- болезни обмена веществ, вызванные нарушением последовательности «букв» генетического кода в отдельных генах;
- митохондриальные заболевания, связанные с нарушениями генетического материала митохондрий;
- низкий процент мозаицизма, когда клеток с неправильным кариотипом очень мало;
- мутации в отдельных генах, не приводящие к изменению внешнего вида хромосом;
- эпигенетические нарушения, при которых не изменяется структура хромосом и генов, но изменяется их функция.
Хромосомный микроматричный анализ (ХМА) используется для получения дополнительной информации, не видимой в световой микроскоп. С его помощью можно изучать все клинически значимые участки генома и выявлять изменения числа и структуры хромосом, а именно микроразрывы (микроделеции и микродупликации).
При анализе хромосомных микрочипов используется технология полногеномной амплификации и гибридизации экспериментальных фрагментов ДНК с олигонуклеотидами, нанесенными на микрочип. Проще говоря, исследуемая ДНК сначала копируется для увеличения ее количества, а затем смешивается со специальными ДНК-микрочипами, помогающими выявить различные нарушения.
Этот метод позволяет обнаружить делеции и дупликации сегментов ДНК по всему геному. Разрешение стандартного XMA составляет 100 000 пар оснований — «букв» генетического кода (в некоторых регионах 10 000 п.н.).
С помощью HMA можно определить:
- изменения числа хромосом;
- дупликации и делеции, включая микродупликации и микроделеты;
- отсутствие гетерозиготности: потеря одной из двух копий гена. Это явление имеет важное значение в онкологии, при импринтинговых болезнях (когда активность гена зависит от того, каким он является родителем), аутосомно-рецессивных заболеваниях (связанных с рецессивными генами; о них мы поговорим ниже), близкородственных браках;
- однородительские дисомии, когда в геноме ребенка присутствуют две хромосомы от одного родителя.
Мутации в генах и заболевания, к которым они способны приводить
Мутации – это изменения, происходящие в ДНК как случайным образом, так и под воздействием различных факторов, например химических веществ, ионизирующего излучения. Они могут затрагивать как отдельные «буквы» генетического кода, так и большие участки генома. Мутации происходят постоянно, и это главный двигатель эволюции. Большую часть времени они нейтральны, то есть ни на что не влияют, не приносят вреда и не приносят пользы. В редких случаях происходят полезные мутации — они дают организму какие-то преимущества. Бывают и вредные мутации — из-за них нарушается работа важных белков, наоборот, они возникают довольно часто. Генетические изменения, встречающиеся более чем у 1% людей, называются полиморфизмами: это нормальные и естественные вариации ДНК.
Все внешние признаки и особенности организма, которые человек получает от родителей, передаются через гены. Это важнейшее свойство всех живых организмов называется наследственностью. В зависимости от того, как гены проявляются в тех или иных признаках, их делят на две большие группы.
- Доминантные гены. Проще говоря, эти гены «сильнее». Если в клетках присутствует хотя бы одна копия указанного гена, то проявятся его признаки.
- Рецессивные гены «слабее» доминантных. Если у человека есть одна копия гена, который является доминантным, и одна копия, которая является рецессивной, проявится доминантный признак. А для проявления рецессивного признака нужны две соответствующие копии.

Например, карий цвет глаз у человека является доминирующим. Поэтому у кареглазых родителей чаще рождается кареглазый ребенок. Если у одного из родителей карие глаза, а у другого голубые, то вероятность рождения в этой семье детей с карими глазами также высока. У двух голубоглазых родителей, скорее всего, все дети будут голубоглазыми. Но у кареглазых родителей может быть голубоглазый ребенок, если у них обоих есть рецессивные «гены голубоглазости», и они перейдут к ребенку. Конечно, это упрощенная схема, ведь за цвет глаз отвечает не один, а несколько генов, но на практике эти законы наследования часто работают. Точно так же наследственные заболевания могут передаваться потомству.
В ряде случаев у детей и взрослых встречается так называемый мозаицизм. При этом заболевании одна часть клеток имеет нормальный набор хромосом, а другая часть имеет измененный набор. Например, больные мозаичным синдромом Дауна могут иметь довольно гармоничный вид и практически неотличимы от здоровых людей.
Что такое синдром Эдвардса
Синдром Эдвардса или трисомия X Трисомия Трисомия означает вариант хромосомной мутации, при котором клетки человека содержат не 46, а 47 хромосом на 18-й хромосоме — это генетическое заболевание, при котором ребенок наследует три 18-х хромосомы вместо двух. Мутация возникает в результате нерасхождения хромосомы 18 во время деления клетки.
В описаниях болезни говорится, что в 60% случаев из-за дефектов, несовместимых с жизнью, дети с синдромом Эдвардса погибают в утробе матери. В группе трисомий только в трех случаях — при синдроме Эдвардса, синдроме Дауна (трисомия по 21 хромосоме) и синдроме Патау (трисомия по 13 хромосоме) возможно рождение живого ребенка и его дальнейшее развитие, хотя и осложненное. При других вариантах дополнительных хромосом патология несовместима с жизнью.
Распространенность синдрома Эдвардса в мире составляет один случай на 3000-7000 новорожденных, специфической зависимости по регионам или расам нет. Синдром чаще встречается у девочек. Ребенок с трисомией 18 может родиться у любой женщины, но считается, что риск увеличивается с возрастом матери. И некоторые исследования показали, что синдром Эдвардса имеет тенденцию к увеличению в последние годы. Возможно, это связано с улучшением диагностики и увеличением возраста рожениц.
Хотя основные симптомы заболевания были описаны в начале 20 века, первый полный обзор синдрома и его основной причины — появления дополнительной 18-й хромосомы — был проведен только в 1960 году британским медицинским генетиком Джоном. Эдвардс, в честь которого была названа эта патология.
Заболевание сопровождается многочисленными нарушениями в развитии систем органов, но если лишняя 18 хромосома есть не в каждой клетке организма, что встречается у 5% людей с данной патологией и называется мозаицизмом, то синдром Эдвардса может протекать в более легкой форме.
Как и другие заболевания с хромосомными мутациями, синдром Эдвардса неизлечим. В настоящее время лечение состоит в основном из паллиативной помощи.
Как диагностируют синдром Эдвардса
Заподозрить наличие синдрома Эдвардса на УЗИ плода и УЗДГ можно по косвенным признакам: недоразвитие одной из пупочных артерий, многоводие и уменьшение размеров плаценты. Но УЗИ может показать только характерные признаки патологии, оно не может подтвердить наличие именно этого синдрома.

Эпоха Александры Евгений Глаголев, отец тяжелобольной девочки, рассказывает о том, как изменилась жизнь его семьи, почему он решил сменить профессию и почему родителям нужно время от времени куда-то вместе ездить
В целом для диагностики заболевания большое значение имеет совокупность факторов: результаты лабораторного исследования, данные УЗИ, срок беременности и возраст матери. При наличии сразу нескольких симптомов необходимы дополнительные инвазивные исследования: биопсия ворсин хориона (анализ клеток эмбриональной ткани), амниоцентез (забор околоплодных вод на анализ) или кордоцентез (забор крови из пуповины). Полученный в ходе процедур фетальный материал направляют на кариотипирование – анализ на нарушения хромосомного набора.
У новорожденного также следует провести хромосомное исследование с помощью анализа крови, чтобы установить точную причину врожденных дефектов.
Для изучения кариотипа используют специальный метод — световую микроскопию дифференциально окрашенных метафазных хромосом культивируемых лимфоцитов периферической крови.
Как выявляют рецессивные мутации?
Для выявления мутаций, передающихся рецессивно, используют несколько тестов.
Секвенирование по Сэнгеру — это метод секвенирования (определение последовательности нуклеотидов, буквально «чтение» генетического кода) ДНК, также известный как метод обрыва цепи. Анализ используется для подтверждения выявленных мутаций. Это лучший метод для выявления коротких тандемных повторов и секвенирования отдельных генов. Этот метод может обрабатывать только относительно короткие последовательности ДНК (до 300–1000 пар оснований) за раз. Однако самым большим недостатком этого метода является большое количество времени, необходимое для его проведения.
Если неизвестно, какую мутацию необходимо выявить, используются специальные панели.
Исследовательская группа: тестирование на наличие определенных мутаций, перечисленных специальной исследовательской группой. Анализ позволяет одновременно выявить разные мутации, которые могут привести к генетическим заболеваниям. Анализ позволяет сортировать мутации в панели по частоте встречаемости (панели скрининга, предназначенные для выявления носителей патологической мутации, часто встречающейся в определенном регионе или в определенной закрытой популяции) и по пораженным органам или системам органов (панель «Патология соединительной ткани»). Но этот анализ также имеет ограничения. Анализ не выявляет хромосомных аберраций, мозаицизма и мутаций, не вошедших в панель, митохондриальных заболеваний и эпигенетических нарушений.
Не все семьи могут проследить все возможные рецессивные заболевания. Тогда на помощь приходит экзомное секвенирование — тест для определения генетических повреждений (мутаций) в ДНК путем изучения в одном тесте практически всех участков генома, кодирующих белки, изменения которых являются причиной наследственных заболеваний.
Секвенирование следующего поколения-NGS представляет собой определение последовательности нуклеотидов в геномной ДНК или агрегате матричной РНК (транскриптоме) путем амплификации (копирования) множества коротких участков генов. Это разнообразие генных фрагментов в конечном итоге покрывает всю популяцию генов-мишеней или, при необходимости, весь геном.

Анализ выявляет точечные мутации, вставки, делеции, инверсии и пермутации в экзоме. Анализ не выявляет больших корректировок; мутации числа копий (CNV); мутации, вовлеченные в триаллельное наследование; мутации в митохондриальном геноме; эпигенетические эффекты; большие тринуклеотидные повторы; Рецессивные мутации, связанные с Х-хромосомой, могут не распознаваться у женщин с заболеваниями, связанными с неравной инактивацией Х, однородительскими фенокопиями и дисомиями, а также генами, имеющими псевдогены, сходные по структуре.
Что делать, если в семье есть наследственное заболевание?
Есть два способа обнаружить унаследованные генетические мутации у эмбриона:
Преимплантационное генетическое тестирование (ПГТ) в цикле ЭКО. Это диагностика генетических заболеваний у человеческого эмбриона до имплантации в слизистую оболочку матки, то есть до наступления беременности. Обычно для анализа берут биопсию бластомера (эмбриональной клетки) эмбриона на стадии дробления (от 4 до 10 бластомеров). Различают несколько видов ПГТ: при хромосомных аномалиях, при моногенных заболеваниях и при структурных хромосомных перестройках. Данные Simon et al (2018) свидетельствуют о том, что в случае ЭКО с ПГТ у пациентки в возрасте 38-40 лет показатель успешности ЭКО составляет 60%. Но есть ряд ограничений в изучении эмбриона. Поэтому из-за ограниченного числа клеток мозаицизм определить невозможно.
Если ЭКО с ПГТ невозможно, используют второй вариант — исследование плодного материала во время беременности.
Для забора плодного материала используют инвазивные методы:
- биопсия хориона – при взятии клеток из плаценты;
- амниоцентез – когда берут клетки из амниотической жидкости.
Затем эти клетки исследуются с помощью одного или нескольких генетических тестов (которые имеют свои ограничения). Выполнение инвазивных методов может быть связано с риском беременности порядка 1%.
Поэтому путем проведения дополнительных исследований можно значительно снизить риск рождения ребенка с генетическим заболеванием в конкретной семье. Но свести этот риск к нулю сегодня, к сожалению, невозможно, так как любой генетический тест имеет ряд ограничений, что делает невозможным абсолютное исключение всех генетических заболеваний.

Автор статьи
Пелина Ангелина Георгиевна
Проводит генетическое обследование доноров Репробанка, подбирает доноров для пар, у которых ранее были дети с установленной генетической патологией.
Генетические аномалии широко распространены. Об этом свидетельствует тот факт, что 48% доноров полового материала элиминированы имеющимися мутациями, которые могут привести к рождению больных детей. Кроме того, у 30% из них есть серьезные хромосомные патологии, о которых они не знали.
Немного о РНК
Последовательность трех азотистых оснований в мРНК указывает на включение определенной аминокислоты в последовательность белка. Молекулы тРНК переносят эти аминокислоты к рибосомам, где синтезируется белок.
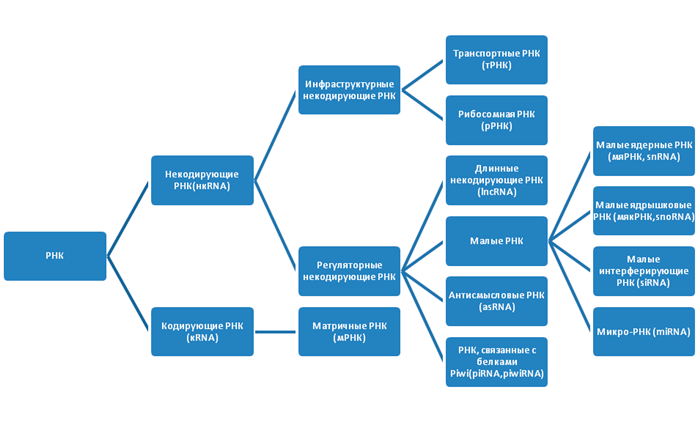
Рисунок 2. Типы РНК
Регуляторные нкРНК очень широко распространены в организме, классифицируются по размеру и выполняют ряд важных функций (табл. 1).
| Имя | Обозначение | Длина | Функции | |
|---|---|---|---|---|
| Длинная некодирующая РНК | днкРНК, днкРНК | 200 нуклеотидов | 1. Регулировать селективное метилирование ДНК, направляя ДНК-метилтрансферазу 2. Управление выборочной настройкой репрессорных комплексов Polycomb |
|
| Малая РНК | Малая ядерная РНК | мяРНК, мяРНК | 150 нуклеотидов | 1. Участвовать в сплайсинге 2. Регулировать активность факторов транскрипции 3. Поддерживать целостность теломер 13 |
| Малые ядрышковые РНК | мякРНК, мякРНК | 60–300 нуклеотидов | 1. Участвовать в химической модификации рРНК, тРНК и яРНК 2. Возможно, участвует в стабилизации структуры рРНК и защите от действия гидролаз |
|
| Малые интерферирующие РНК | миРНК, миРНК | 21–22 нуклеотида | 1. Провести противовирусную иммунную защиту 2. Подавить активность собственных генов |
|
| Микро-РНК | микроРНК, микроРНК | 18–25 нуклеотидов | Подавить трансляцию с помощью РНК-интерференции | |
| Антисмысловая РНК 14 | asРНК | 1. Короткие: менее 200 нуклеотидов 2. Длинный: более 200 нуклеотидов |
Блокировать трансляцию, образуя гибриды с мРНК | |
| РНК, связанная с белками Piwi | пиРНК, пивиРНК | 26–32 нуклеотида | Также называемые «хранителями генома», они подавляют активность мобильных генетических элементов во время эмбриогенеза | |
Проблема терминологии
Прежде чем ответить на вопрос: «Сколько у нас генов?», нам нужно понять, что такое ген?
Основное внимание HGP уделялось генам, кодирующим белок 15. Однако, как указано в исходном отчете HGP в 2001 г., «тысячи генов человека производят некодирующую РНК (нкРНК) в качестве конечного продукта», хотя в то время около 706 г. Были известны гены нкРНК 2. В своей недавней статье, опубликованной в журнале BMC Biology, Стивен Л. Зальцберг определяет ген 16 следующим образом:
Ген — это любой сегмент хромосомной ДНК, который либо транскрибируется в функциональную молекулу РНК, либо сначала транскрибируется в РНК, а затем транслируется в функциональный белок.
Это определение включает как гены некодирующей РНК, так и гены, кодирующие белок, и позволяет определить все альтернативные варианты сплайсинга в одном локусе как варианты одного и того же гена. Это позволяет исключить псевдогены, нефункциональные остатки структурных генов, утративших способность кодировать белок.
Результаты первых двух исследований указывали на наличие у человека 31 0002 и 26 588 белок-кодирующих генов17, а в 2004 г появилась полная последовательность генома человека4, и авторы подсчитали, что полный каталог содержит 24 000 белок-кодирующих генов. Каталог генов человека Ensembl включает 22 287 генов, кодирующих белки, и 34 214 транскриптов.
На самом деле обезьян всегда называли нашими ближайшими родственниками. Сейчас наука пытается доказать, что семейство свиней нам ближе всего. Однако это не означает, что человек произошел от свиньи.
Предрасположенность = болезнь?
— Нормальная структура гена обеспечивает определенную функцию или признак у человека. В данном случае речь идет о полном здоровье.
Однако бывают ситуации, когда человек предрасположен к определенным нарушениям здоровья. Но это не значит, что вы обязательно заболеете. Речь идет о полиморфизме (варианте нормальной структуры гена). В этой ситуации болезнь будет проявляться под влиянием факторов риска. Например, для гипертонии это будут: ожирение, малоподвижный образ жизни, курение и т.д.
Если обнаруживается мутация в структуре гена, это приводит к изменению функции гена и, как следствие, к патологии. В генетике такие виды болезней называются «моногенными болезнями», среди них есть рецессивные.

Давайте посмотрим, как это работает:
- если каждый из родителей «отдает» будущему потомству здоровый ген, рождается здоровый малыш;
- если один из родителей дает ген с мутацией, а второй — здоровый, рождается здоровый рецессивный носитель. Это означает, что сам ребенок будет здоров на протяжении всей своей жизни, но по-прежнему будет носителем генетического заболевания и в дальнейшем может передать его своим детям;
- если оба гена от матери и отца имеют мутации, ребенок рождается с заболеванием.
Каждая пара находится в группе риска, но еще на этапе планирования беременности они могут пройти молекулярно-генетическое исследование и уточнить свой генетический статус.
Современные молекулярно-генетические технологии позволяют определять полиморфизмы и (или) мутации несколькими способами:
- в определенном месте гена;
- анализировать всю структуру гена;
- одновременно тестировать группу генов (например, для выявления нервно-мышечных заболеваний).
Как паре, планирующей ребенка, понять, что нужно идти к врачу-генетику?
1 вариант При отсутствии видимых причин дальнейшее генетическое тестирование не требуется. Если терапевт, гинеколог или другой специалист выявляет какие-либо особенности в родословной, состоянии здоровья или беременности, то женщина (или супружеская пара) направляется на консультацию к генетику, который разрабатывает индивидуальный план генетического обследования.
вариант 2. Пары, не имеющие проблем со здоровьем, могут пройти генетическое тестирование самостоятельно по своему усмотрению. Например, прогностические тесты на наследственную предрасположенность к некоторым заболеваниям. То есть сделать так называемый «генетический паспорт». Благодаря этому скринингу люди могут узнать предрасположенность, например, к артериальной гипертензии, тромбозам, бронхиальной астме или изменению физиологического течения беременности и заранее предупредить эти заболевания или осложнения.

Еще один вид исследования, который можно планировать самостоятельно, — скрининг перед зачатием на носительство рецессивных мутаций. Тесты позволяют оценить риск рождения ребенка с наследственным заболеванием у конкретной пары. Территориальные сообщества имеют свои особенности генофонда. В каждом условно замкнутом сообществе накапливаются свои рецессивные мутации.
Естественно, что вероятность встречи двух партнеров (супругов) с патогенными мутациями в одних и тех же генах крайне мала. Однако хуже, если родители узнают о своих генотипических особенностях слишком поздно, когда у них уже есть тяжелобольной ребенок. Вот для чего нужен скрининг преконцепции.
Если по результатам исследования оба потенциальных родителя являются здоровыми носителями мутаций в одних и тех же генах, риск рождения больного ребенка будет высоким (25%). В этом случае при беременности рекомендуется пренатальная молекулярно-генетическая диагностика для уточнения, благоприятный или неблагоприятный вариант унаследовал плод.
Анализы позволяют понять, будет ли плод больным, здоровым или здоровым носителем. И в зависимости от результатов врачи и будущие родители принимают решение о сохранении или прерывании беременности больным плодом. Окончательное решение остается за семьей.

Родители не могли в это поверить, ведь сами были здоровы и уже имели совершенно здорового ребенка, но врачи объяснили, что дети с генетическими проблемами иногда появляются из-за спонтанных мутаций в генах или хромосомах, а не только наследственности.
Как расшифровать результат анализа
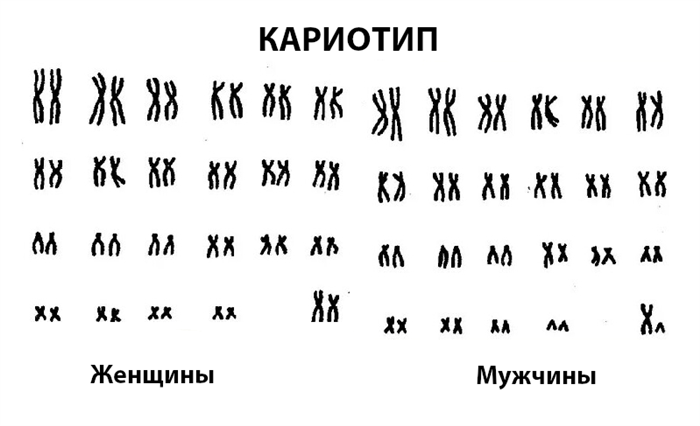
Нормальный хромосомный набор человека содержит 46 хромосом, 2 из которых являются половыми хромосомами: XX или XY. Поэтому нормальный набор хромосом здорового человека обозначается:
Для удобства каждой хромосоме присвоен определенный номер. Это нужно для того, чтобы в дальнейшем стало понятно, где дефект. Длинное плечо хромосомы обозначается буквой q, короткое плечо – t.
Так, например, дефект на хромосоме 5 у самки обозначается 46ХХ5т, где 46ХХ указывает на женский набор хромосом, а обозначение 5т указывает на то, что пострадало короткое плечо хромосомы 5. Эта генетическая аномалия называется синдромом плачущей кошки. У больных детей особое строение гортани, поэтому они не плачут, а издают звуки, похожие на кошачье мурлыканье. Заболевание сопровождается изменением черт лица и умственной отсталостью.
Что делать, если хромосомный анализ показал вероятность рождения больного ребенка
Все зависит от конкретного заболевания, вероятности наследственности и отношения к этому вопросу будущих родителей. Но в любом случае такой паре необходима консультация репродуктолога.
Некоторые пары все же решаются на риск и заводят ребенка, не прибегая к репродуктивным технологиям. В этом случае они ценят важность принятого решения и рождение особенного малыша не станет для них неожиданностью.
Более разумным решением является ЭКО с предимплантационной диагностикой. В этом случае зачатие происходит вне организма женщины, эмбрионы проверяются на генетику, а затем подсаживаются в матку.
Так как кариотип не меняется на протяжении всей жизни, достаточно один раз пройти такой анализ. Изучение кариотипа лимфоцитов периферической крови позволит узнать их хромосомные особенности и свести к минимуму риск заболевания ребенка.